Finding new materials for clean-energy chemical storage and separation constitute one of the challenges in fighting climate change, and a combination of large-scale physical simulations with machine learning methods has dramatically accelerated the materials discovery processes. Although machine learning has shown great success in various fields, incorporating physical information into machine learning methods is crucial to improving their predictive performance. My research focuses on employing physical principles governing the storage and separation behavior of materials to develop new machine learning techniques on large datasets generated by simulations of these materials.
The PDF file of my full research description can be downloaded here.
See here for my full list of publications.
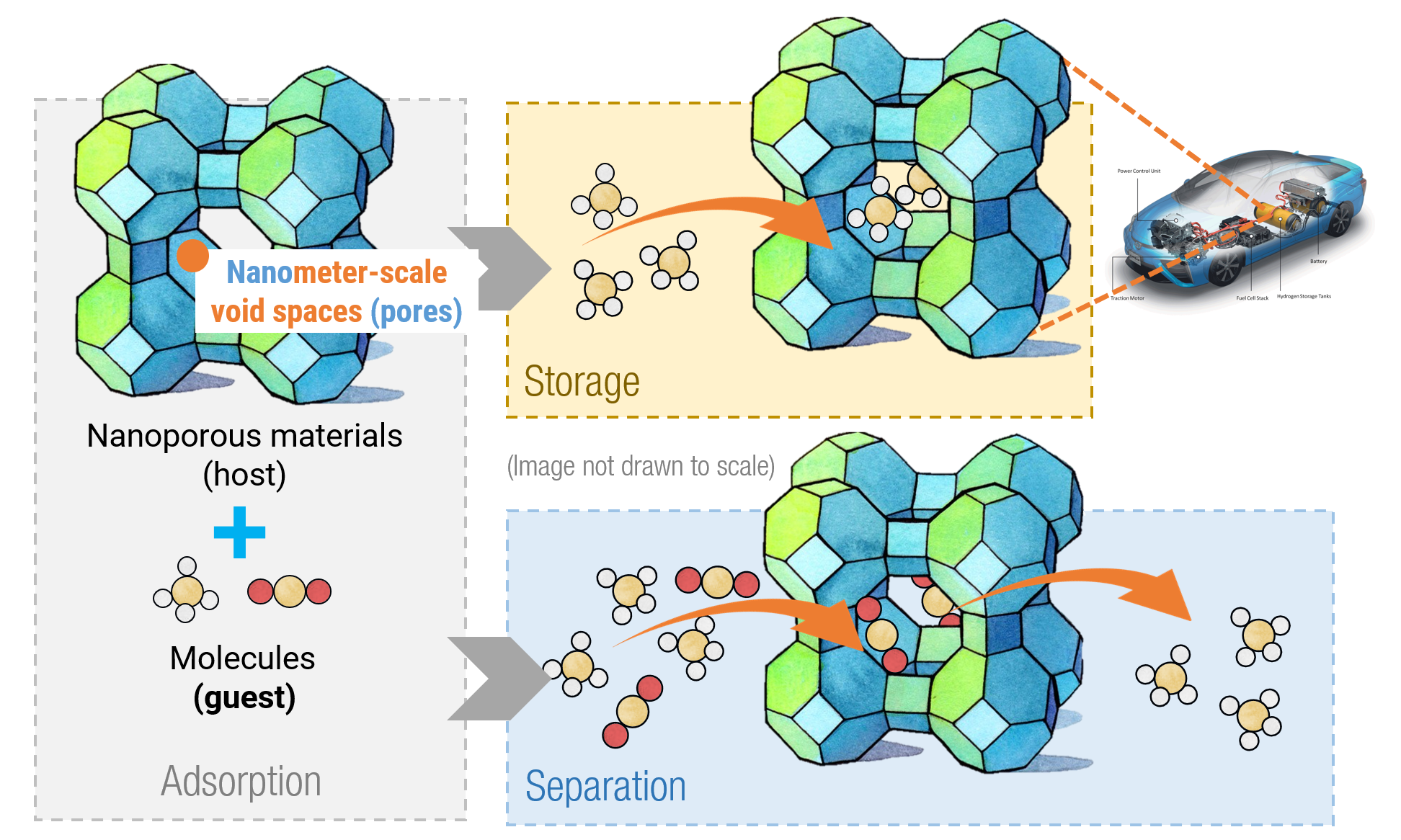
The physical systems studied in my research mainly involve nanoporous materials, a type of materials which contain void spaces in the size of a few to tens of nanometers. These void spaces can act as “rooms” to contain various types of small molecules, and such phenomena of nanoporous materials encapsulating guest molecules are called adsorption.By designing the structure of nanoporous materials towards favorable adsorption properties, they can achieve exceptional storage capacities for important molecules or high separation efficiencies for chemical mixtures.
Deep neural network learning of complex binary sorption equilibria from molecular simulation data
[Paper] [Code] [Cover Art]
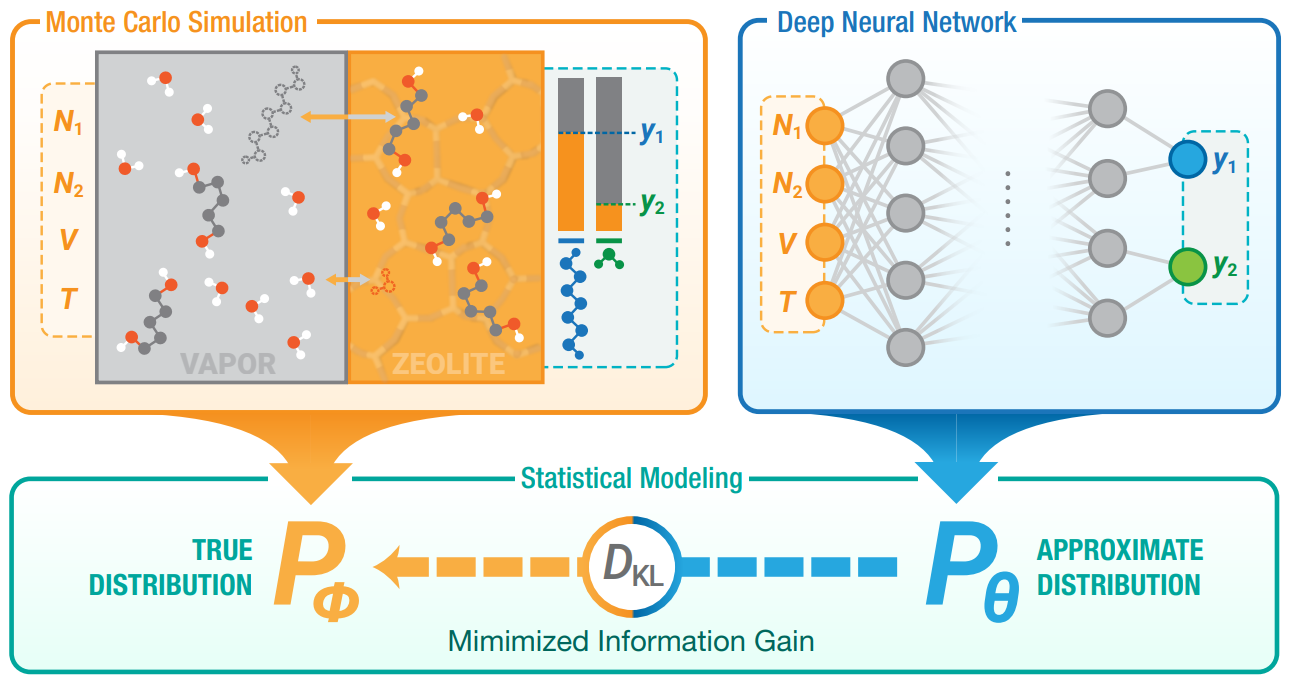
We developed a learning algorithm which minimizes the KL divergence between the “true” statistical mechanics distribution and the approximating distribution parametrized by a neural network. In complex adsorption systems where domain-specific models fail to predict the simulation results, a neural network trained in such manner was able to accurately approximate the simulations to the same magnitude as their precision. The predictions from the neural network was utilized to facilitate optimization the conditions of a desorptive drying process, which requires a laborious iterative search if undertaken by simulation alone. Furthermore, it allows for application of transfer learning by fine-tuning a pretrained network for a different chemical system.
Fingerprinting nanoporous materials for hydrogen storage using meta-learning
[NeurIPS Workshop Paper] [Poster] [Code]
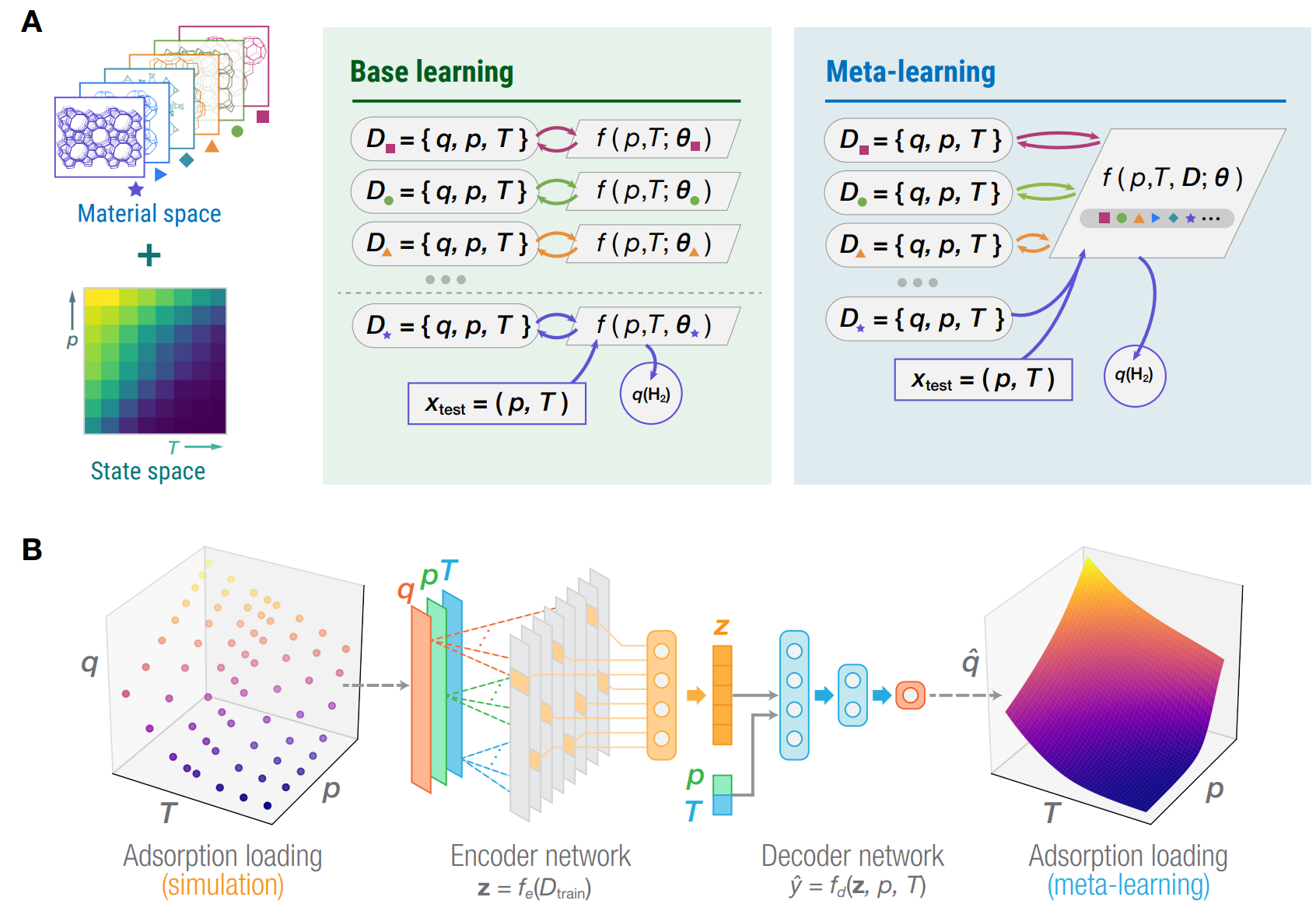
Adsorption using nanoporous materials is one of the emerging technologies for hydrogen storage in fuel cell vehicles, and efficiently identifying the optimal storage temperature requires modeling hydrogen loading as a continuous function of pressure and temperature. Using data obtained from high-throughput Monte Carlo simulations for diverse nanoporous materials, we develop a meta-learning model which jointly predicts the adsorption loading for multiple materials over wide ranges of pressure and temperature. Meta-learning gives higher accuracy and improved generalization compared to fitting a model separately to each material. Here, we apply the meta-learning model to identify the optimal hydrogen storage temperature with the highest working capacity for a given pressure difference. Materials with high optimal temperatures are found closer in the fingerprint space and exhibit high isosteric heats of adsorption. Our method and results provide new guidelines toward the design of hydrogen storage materials and a new route to incorporate machine learning into high-throughput materials discovery.
Interpretable Learning of Complex Multicomponent Adsorption Equilibria from Self-attention
[Paper] [Poster]
![[Poster]](/assets/poster_ML4Molecules_2020_18.png){kind=link}
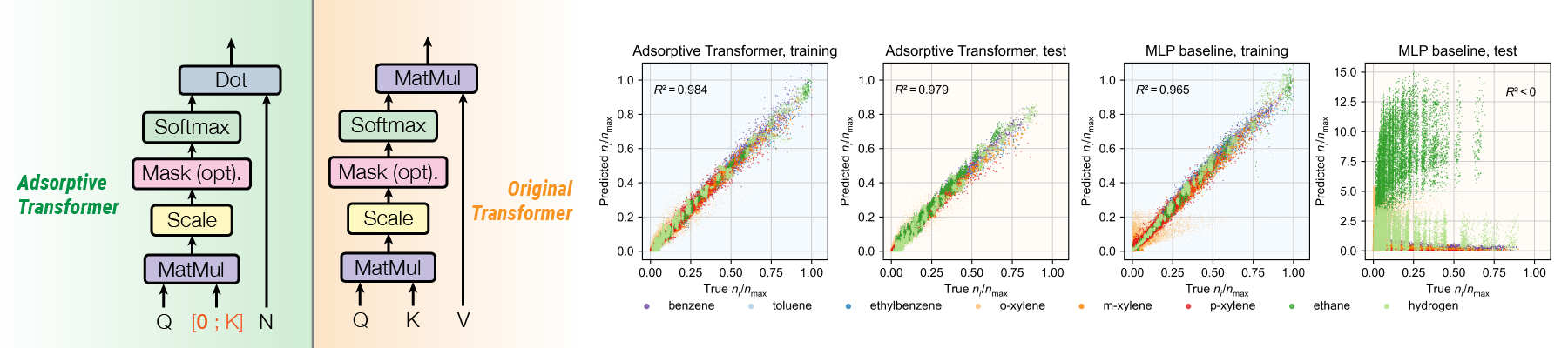
Modeling complex interactions in multicomponent adsorption equilibria is pivotal for the development of efficient chemical separation processes using nanoporous materials. Starting from the ideal-mixture adsorption model in statistical thermodynamics, we developed a machine learning formulation of multicomponent adsorption, Adsorptive Transformer, based on the self-attention mechanism. The Adsorptive Transformer accounts for the effects of guest-guest interactions, and a “shallow” model is already able to significantly outperform a deep neural network on extrapolation in the state space. The model is also readily interpretable to show adsorption mechanisms, such as cooperative adsorption and sieving.